DNA Sequencing –is a biochemical method to figure out the arrangement of A, T, G and C bases in a DNA molecule. Every specific arrangement of letters (code) in a piece of DNA means something to the cell and therefore our body. For example, the DNA sequence of a gene tells us the specific code that makes up that gene and what it does.
The ability to sequence DNA got a boost in 1975 when a scientist named Fred Sanger figured out an easy way as compared to earlier methods. As explained in the structure of DNA (see DNA), the fact is that DNA is double stranded, it is negatively charged and it is also directional. In other words, DNA is organized on one strand in one direction and the other strand (called the complementary strand) in the opposite direction. This is shown in Figure 3A below.
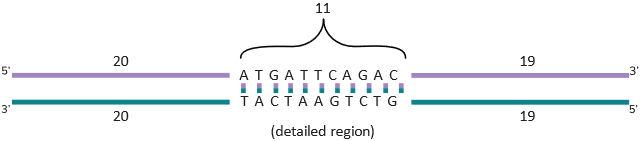
In this figure, a small section of the DNA is shown in detail with the sequence ATGATTCAGAC written in the standard way DNA sequence is shown, that is, in the 5’ to 3’ direction. So it will be written 5’-ATGATTCAGAC-3’. As shown in the figure, you could have sequence before it and after it, but this is how sequence information is presented in an acceptable format. You also know that A always pairs with T and G always pairs with C. This is indicated by the lines drawn between them. With this, you can see that if you use one strand, you can figure out the other strand as it is always going to be complementary. In other words, the opposite strand sequence of the highlighted sequence in Figure 3A is 5’ -GTCTGAATCAT-3’. As you can see, it is written always in the 5’ to 3’ direction though Figure 3A shows the bottom strand as 3’ -TACTAAGTCTG-5’. If you look at the end of the sequence and try to read it from the 5’ end, you will see the correct sequence as written before (GTCTGAATCAT).
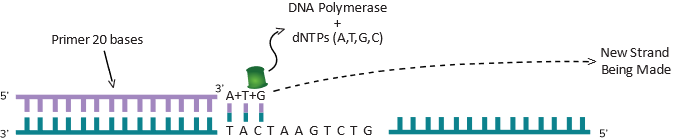
As mentioned in the DNA section, two strands of DNA (complementary strands) make a complete double helix molecule of DNA. The two strands can be completely separated into single strands by heating to a temperature close to 100oC. If you cool the tube, the two strands that are complementary will find each other and come back to form the original molecule. Inside a living cell, there is an enzyme (protein) called DNA Polymerase that can copy DNA and that is how DNA is copied or replicated in the cell. This enzyme is able to bind to one strand of DNA and using that strand, add bases that are complementary and then keep on going until it copies one strand completely. As shown below in Figure 3B, this polymerase moves in one direction using the strand of DNA that is going in the opposite direction.
As you can imagine, another polymerase enzyme coming from the other side (using the other strand), can do the same thing (shown in Figure 3C) and now you can have both strands copied.
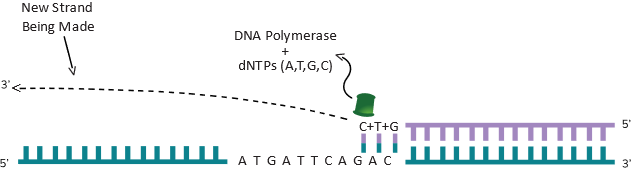
Essentially, you have created a copy of the original piece of DNA. Assume that this piece of DNA is 50 bp long and there is one molecule as shown in the Figure 3A You can make a lot of copies of this DNA in a small plastic reaction tube. For the polymerase to start copying DNA in a tube, all it needs is a starting point marked as a Primer as shown in the Figure 3B. Like any piece of DNA, the primer will also have a 5’ and 3’ end and once the polymerase sees a 3’ end, it knows it should start copying DNA from that end. The role of the primer is to “prime” the synthesis of DNA. To copy DNA, all you have to do is to separate the two strands of DNA (heat can do that), provide a primer, the enzyme DNA polymerase and the four nucleotides dATP, dTTP, dGTP and dCTP. They are the A, T, G and C bases that are attached to a sugar molecule and hence called nucleotides. Technically they are called dATP (deoxy adenosine tri-phosphate) dTTP (deoxy thymine tri-phosphate), dGTP (deoxy guanosine tri-phosphate) and dCTP (deoxy cytosine tri-phosphate). The A, T, G and C has to be in this deoxy tri-phosphate form for the polymerase to synthesize DNA. Under this scenario, the DNA polymerase will copy the complementary base when it finds it. In other words, as shown in Figure 3B, when it sees a T in its path, it will now put the complementary A, when it sees a A (next base in the figure), it will put a T and when it sees a C, it will put a G as the next base and so on. This way, whatever the size of the DNA (50 bases long in this simple example), the enzyme will copy in one direction until the 50th base. As shown in Figure 3C, if you have a primer coming in the other direction, it will copy the DNA till the 50th base also and stop. So, if you know the DNA sequence beforehand, you can copy any piece of DNA by putting two primers exactly at the distance you want and it will copy everything in between. This is what Figure 3B and 3C are showing. The size could be 1000 base pairs long or it could be 100 base pairs long. We are using a simple example of a 50 bp long fragment of DNA to explain some interesting things to you. For example, you can imagine a continuous cycle of opening the two strand of DNA (by heating), cooling the DNA down (allows the primers to find the right spot to sit down) and if that happens, the polymerase will see the 3’ end of the primers (each time it cools down) and make a piece of DNA. Remember, you have two primers going in opposite directions (if you imagine both Figure 3B and 3C happening in the same tube). As it is happening in both directions, as explained before in the example, a 50 base pair piece of DNA will be copied. Each time it is copied, you have doubled the amount of DNA if this process is done in a tube. By the second round, you have 4 molecules of 50 bp made. By the 3rd round, you have 8 molecules and by the 4th round you have 16 molecules and so on. This is called exponential amplification of DNA and also known by another popular term: polymerase chain reaction or PCR. You just learned what PCR is. The primers used in PCR are synthetically made since you are always amplifying things you sort of know the DNA sequence of (at least you know enough to make a primer). Synthetic primers will work inside a reaction done in a tube in a lab. In living things, there is a different system at work which we will not discuss here. By the way, the idea behind PCR won a Nobel Prize in 1987 for a scientist called Kari Mullis. To learn more about PCR, you can visit this site: https://www.genome.gov/genetics-glossary/Polymerase-Chain-Reaction
We went on a little side story here to show how DNA can be copied and how that idea was used for sequencing is where we are going back to now. PCR was not known when Professor Sanger figured out DNA sequencing, but he knew how the polymerase enzyme worked. He also figured out that if he uses certain nucleotides that cannot be extended (or added on) by the polymerase enzyme, it will stop where the non-extendable nucleotide was added. These non-extendable nucleotides are called “terminator” nucleotides. These terminator nucleotides are similar to the normal nucleotides (bases) that make up DNA but have a small piece missing which prevents the next base from being added. They are called dideoxy nucleotides. So, the A terminator is called dideoxy A (ddATP) and the G terminator is called dideoxy G (ddGTP), the T terminator is called dideoxy T (ddTTP) and the C terminator is called dideoxy C (ddCTP). What Fred Sanger figured out is that he can use the normal A, T, G and C nucleotides (dATP, dTTP, dGTP and dCTP) as well as a small amount of one of the terminators and whenever the polymerase runs into that terminator (at the right spot) it will stop. As shown below, here is what will happen if a reaction with all 4 of the normal nucleotides (dNTPs or dexoy nucleotide tri-phosphates to indicate that all 4 are there) and just the C terminator (ddCTP) was used. Remember, only a small fraction of the dCTP in the reaction is replaced with the terminator ddCTP. As you can see in the example below (see Figure 3D) the first G is the seventh base from the 3’ end of the primer. Assume this artificial primer is 20 bases long. The polymerase will copy the DNA and get to the 7th position and it could put a normal dCTP (complementary to the G) and move on to the 8th position and put a dATP and keep going. The next time it comes back to make another copy, it could put ddCTP at position 7 and this time it will stop.
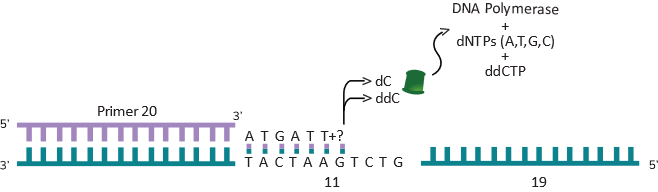
Similarly, it will go around another time and put a ddCTP at position 11 and stop (see Figure 3E below).
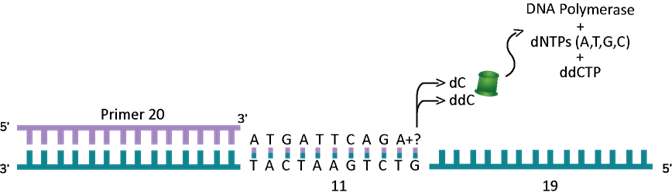
If you add the number of bases from the end of the primer used, the position 7 is actually 27 bases from the primer (which is 20 bases long). So, if you look at the new piece of DNA made by the polymerase, where the ddCTP was put at position 7 is now 27 bases long and the position 11 where a ddCTP was put is 31 bases long. In this reaction, if you can figure out the size of the DNA, you will see two pieces of DNA – one is 27 bases long and the other is 31 bases long. See Figure 3F below for details.
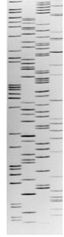
Since you know where the primer started, you know that the 27th position from the 5’ end of the primer is C (or G on the strand that you sequenced) and the 31st position is also a C. In the 1970s, the way to figure this out was to run the sequencing reaction on a very thin DNA separating media called a gel (yes it was a gelatinous material) and the primer was labeled with a radioactive chemical. The gel was used to separate different sizes of DNA molecules by passing electric current through the gel. DNA is a charged molecule and will move through the gel towards the positive pole (see DNA gel in the DNA Section). As shown in the Figure 3G below, you can see that if you just ran the C reaction (as it is called) you will see the following 3 pieces of DNA; primer DNA (at 20 bases long), the first product which is primer plus 7 bases at 27 bases long and the third piece which is primer plus 11 bases which is 31 bases long.
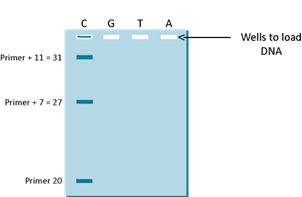
Similarly, if you ran the G reaction, the T reaction and the A reaction (all in separate tubes) and then you load all of the 4 reactions (C, G, T and A) on a gel, you will see the following gel pattern as shown in Figure 3H. The only difference in the reactions is that the C reaction had a small amount of ddCTP, the G reaction had a small amount of ddGTP, the T reaction had a small amount of ddTTP and the A reaction had a small amount of ddATP.
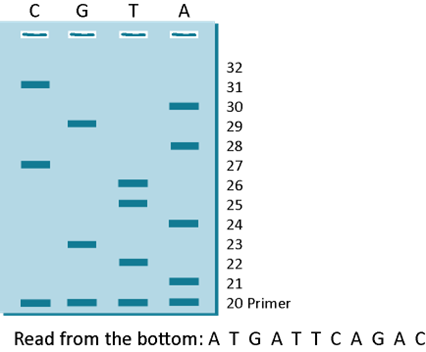
The lanes in this gel were loaded C, G, T and A and so you will read it from the bottom starting at the smallest “band” of DNA from the primer band and then reading the next base in whichever lane the next piece of DNA is. As indicated, the primer band is shown as starting at base 20 because you know that fact. All you have to do is to read the sequence upwards. Try reading the sequence and then write it down and see if it matches 5’-ATGATTCAGAC-3’. To know the sequence of the fragment you sequenced, all you have to do is to flip it and you get the sequence of the strand that was sequenced. In this example, the flipped sequence is 5’-GTCTGAATCAT-3’ and as you can see, it is the exact sequence written on the strand that the polymerase enzyme copied in Figure 3D. This is how Scientists read the sequence of DNA or they did it this way in the 1970s and 1980s. The DNA was labeled with radioactive chemicals and the sequencing reaction was detected as a picture (shown below) after placing an X-ray film on the gel that was run with the A, T, G and C reaction loaded side by side. Once the X-ray film was developed, you got a picture of the gel as shown below in Figure 4 and someone just sat down with a ruler and read the gel from bottom to top like you did before.
This method is called Sanger Sequencing (named after Fred Sanger). Professor Sanger won a Nobel Prize for this in 1980. He won a previous Nobel in 1958 for figuring out the structure of Insulin. More information about Fred Sanger and his sequencing method can be found here: https://dnalc.cshl.edu/view/15479-Sanger-method-of-DNA-sequencing-3D-animation-with-narration.html
In the 1990s, fluorescent dyes were used to label DNA and then images were created from a gel using machines like the LI-COR Sequencer (Figure 5) or the ABI Sequencer (Figure 6) which then converted the bands in a gel into what are known as chromatograms (see the colored graphs – Figure 7). These chromatograms were also read from bottom to top as you would read the gel from an X-ray film.



Then came Next Generation Sequencing (NGS) machines that used similar ideas but instead of gels, used capillary tubes or glass slides to figure out the sequences. Most used the chemistry developed by Professor Sanger or ideas that were modifications. These resulted in machines called SoLID, Ion Torrent, Illumina etc. These machines could sequence billions of bases in a day. For a review of NGS technologies, you can try these two publications:
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9537002/
By the end of 2013, the third-generation machines using single molecule sequencing methods showed up and those include machines made by companies like PacBio and Nanopore Technologies. Today, these machines are widely used in everything from discovery work to diagnosing diseases. For more information on third generation sequencing technologies and their capabilities, you can read these publications.